

Extensive chromosomal rearrangements and rapid evolution of novel effector superfamilies contribute to host adaptation and speciation in the basal ascomycetous fungi
- PMID: 31916390
- PMCID: PMC7036362
- DOI: 10.1111/mpp.12899
Extensive chromosomal rearrangements and rapid evolution of novel effector superfamilies contribute to host adaptation and speciation in the basal ascomycetous fungi
Abstract
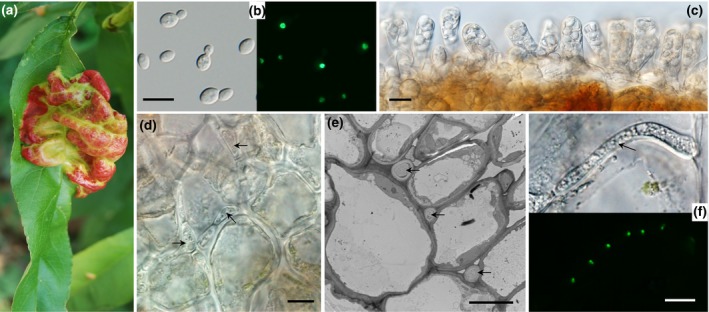
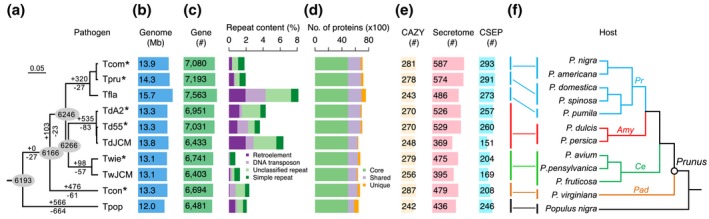
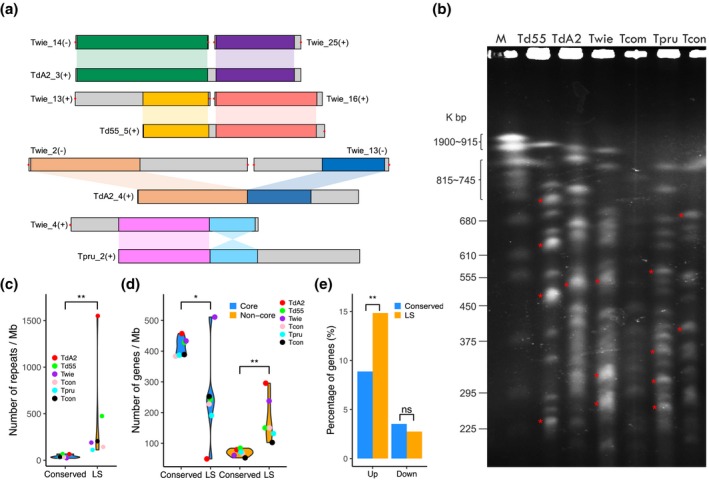
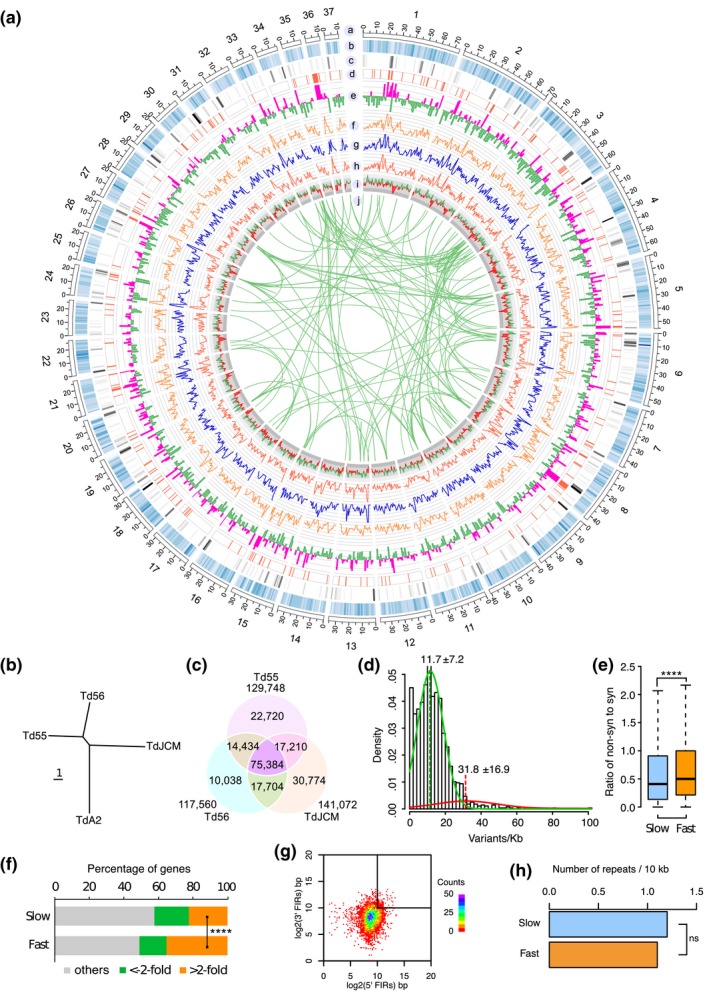
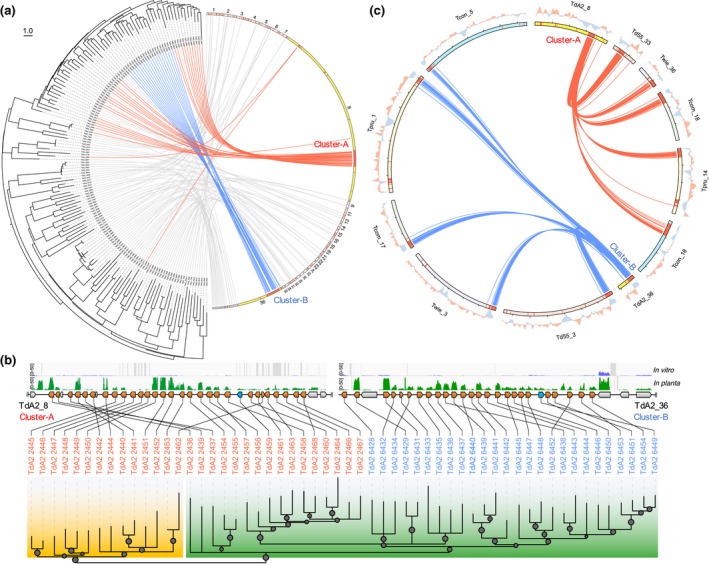
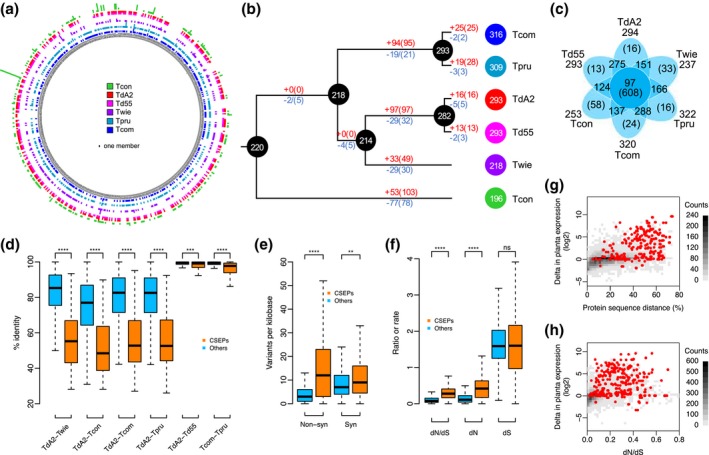
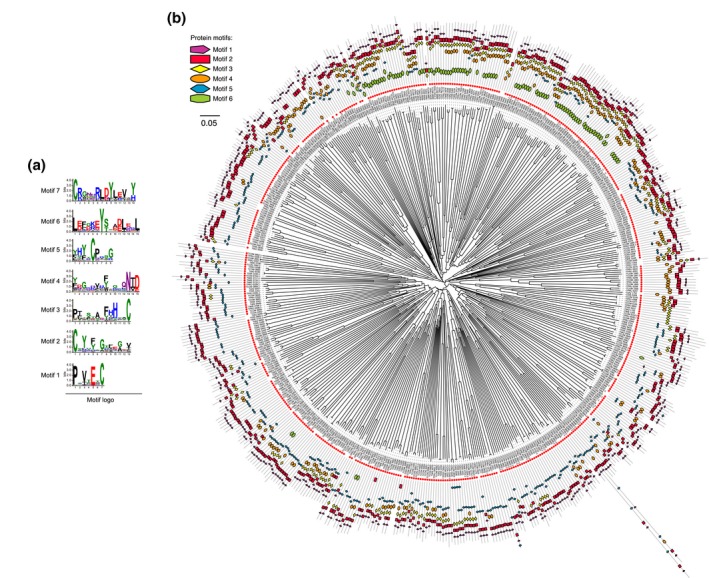
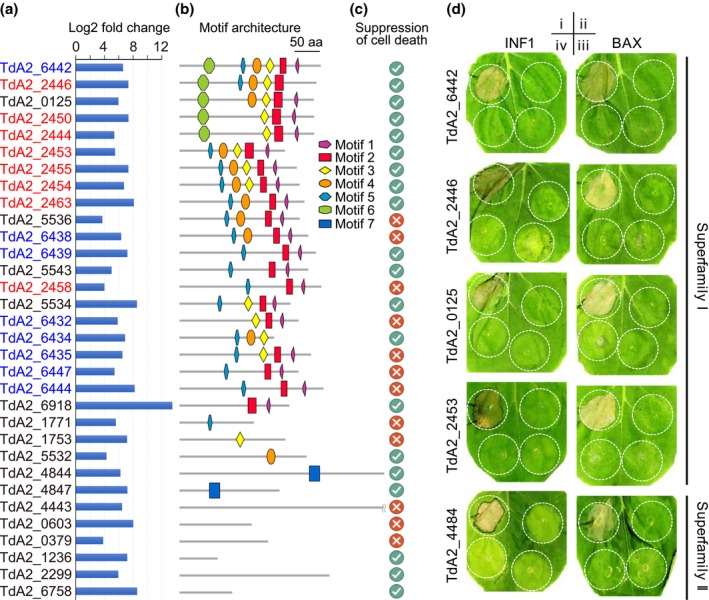
The basal ascomycetes in genus Taphrina have strict host specificity and coevolution with their host plants, making them appealing models for studying the genomic basis of ecological divergence and host adaption. We therefore performed genome sequencing and comparative genomics of different Taphrina species with distinct host ranges to reveal their evolution. We identified frequent chromosomal rearrangements and highly dynamic lineage-specific (LS) genomic regions in Taphrina genomes. The LS regions occur at the flanking regions of chromosomal breakpoints, and are greatly enriched for DNA repeats, non-core genes, and in planta up-regulated genes. Furthermore, we identified hundreds of candidate secreted effector proteins (CSEPs) that are commonly organized in gene clusters that form distinct AT-rich isochore-like regions. Nearly half of the CSEPs constitute two novel superfamilies with modular structures unique to Taphrina. These CSEPs are commonly up-regulated during infection, enriched in the LS regions, evolved faster, and underwent extensive gene gain and loss in different species. In addition to displaying signatures of positive selection, functional characterization of selected CSEP genes confirmed their roles in suppression of plant defence responses. Overall, our results showed that extensive chromosomal rearrangements and rapidly evolving CSEP superfamilies play important roles in speciation and host adaptation in the early-branching ascomycetous fungi.
Keywords: Taphrina; adaptation; comparative genomics; fungi; host specificity; secreted effector protein; speciation.
© 2020 The Authors. Molecular Plant Pathology published by British Society for Plant Pathology and John Wiley & Sons Ltd.
Figures
Similar articles
-
Comparative genome analyses reveal sequence features reflecting distinct modes of host-adaptation between dicot and monocot powdery mildew.BMC Genomics. 2018 Sep 25;19(1):705. doi: 10.1186/s12864-018-5069-z. BMC Genomics. 2018. PMID: 30253736 Free PMC article.
-
Comparative genomics of Taphrina fungi causing varying degrees of tumorous deformity in plants.Genome Biol Evol. 2014 Apr;6(4):861-72. doi: 10.1093/gbe/evu067. Genome Biol Evol. 2014. PMID: 24682155 Free PMC article.
-
Comparative genomics to explore phylogenetic relationship, cryptic sexual potential and host specificity of Rhynchosporium species on grasses.BMC Genomics. 2016 Nov 22;17(1):953. doi: 10.1186/s12864-016-3299-5. BMC Genomics. 2016. PMID: 27875982 Free PMC article.
-
Long-term and rapid evolution in powdery mildew fungi.Mol Ecol. 2024 May;33(10):e16909. doi: 10.1111/mec.16909. Epub 2023 Mar 20. Mol Ecol. 2024. PMID: 36862075 Review.
-
Fungal regulatory evolution: cis and trans in the balance.FEBS Lett. 2009 Dec 17;583(24):3959-65. doi: 10.1016/j.febslet.2009.11.032. FEBS Lett. 2009. PMID: 19914250 Free PMC article. Review.
Cited by
-
Advances in understanding the evolution of fungal genome architecture.F1000Res. 2020 Jul 27;9:F1000 Faculty Rev-776. doi: 10.12688/f1000research.25424.1. eCollection 2020. F1000Res. 2020. PMID: 32765832 Free PMC article. Review.
-
Evolution and molecular bases of reproductive isolation.Curr Opin Genet Dev. 2022 Oct;76:101952. doi: 10.1016/j.gde.2022.101952. Epub 2022 Jul 16. Curr Opin Genet Dev. 2022. PMID: 35849861 Free PMC article. Review.
-
Genomic Instability in Fungal Plant Pathogens.Genes (Basel). 2020 Apr 14;11(4):421. doi: 10.3390/genes11040421. Genes (Basel). 2020. PMID: 32295266 Free PMC article. Review.
-
Enhanced oxidative stress resistance in Ustilago maydis and its implications on the virulence.Int Microbiol. 2024 Oct;27(5):1501-1511. doi: 10.1007/s10123-024-00489-8. Epub 2024 Feb 24. Int Microbiol. 2024. PMID: 38401003 Free PMC article.
-
Telomere-to-Telomere Genome Sequences across a Single Genus Reveal Highly Variable Chromosome Rearrangement Rates but Absolute Stasis of Chromosome Number.J Fungi (Basel). 2022 Jun 25;8(7):670. doi: 10.3390/jof8070670. J Fungi (Basel). 2022. PMID: 35887427 Free PMC article.
References
-
- Agrios, G.N. (2005) Plant Pathology, 5th edition Amsterdam, Netherlands: Academic Press.
-
- Benaglia, T. , Chauveau, D. , Hunter, D.R. and Young, D. S. (2009) mixtools: an R package for analyzing finite mixture models. Journal of Statistical Software, 32, 1–29.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
