

Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis
- PMID: 32049046
- PMCID: PMC7321798
- DOI: 10.1016/j.ccell.2020.01.001
Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis
Abstract
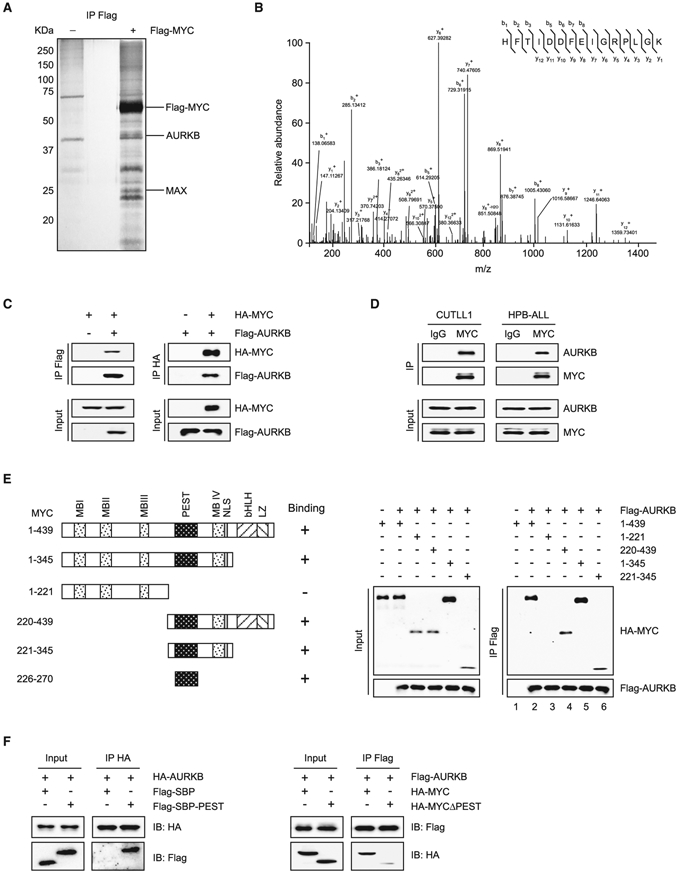
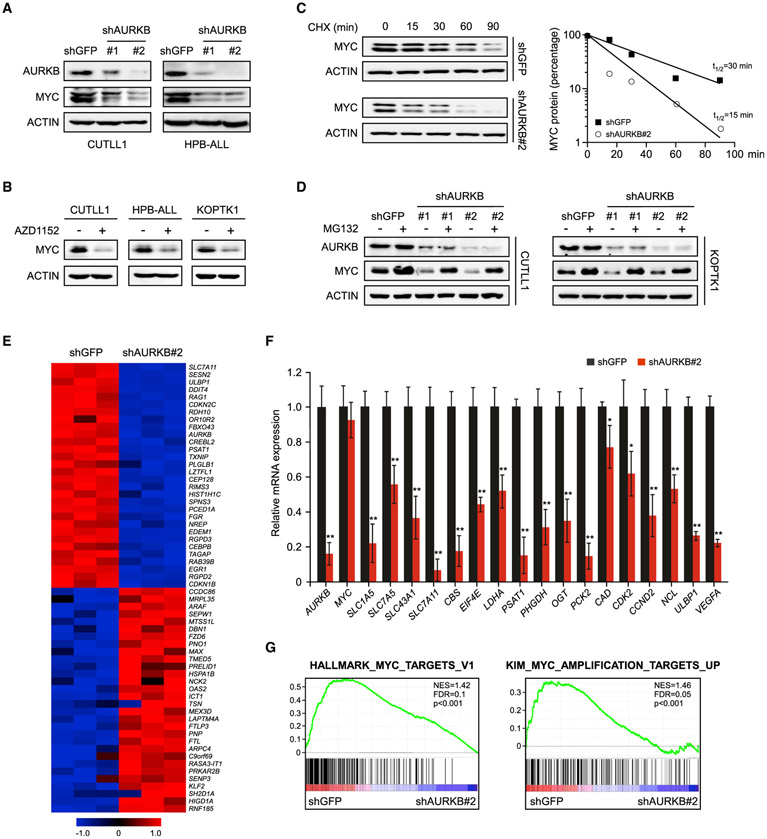
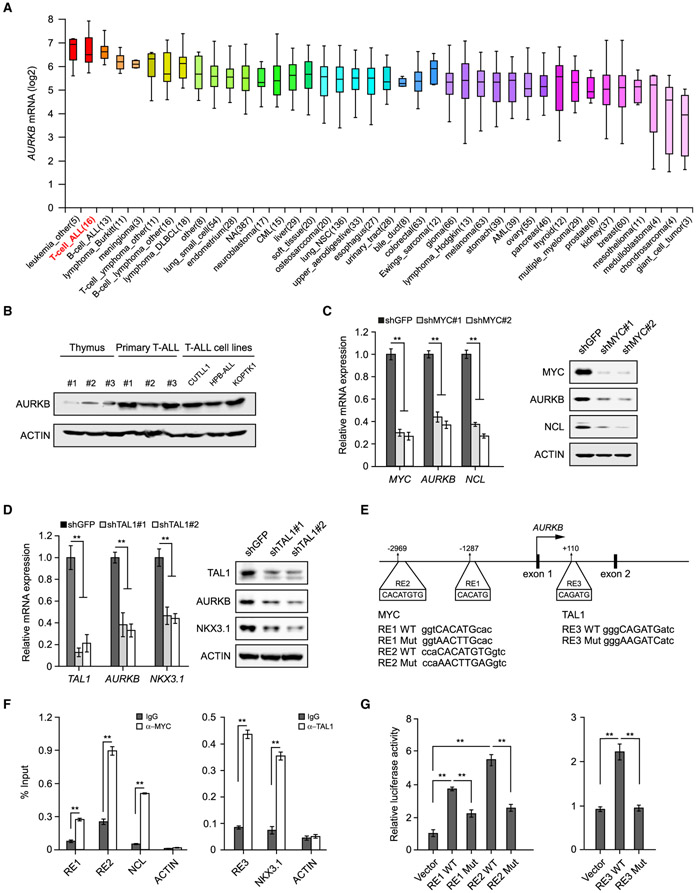
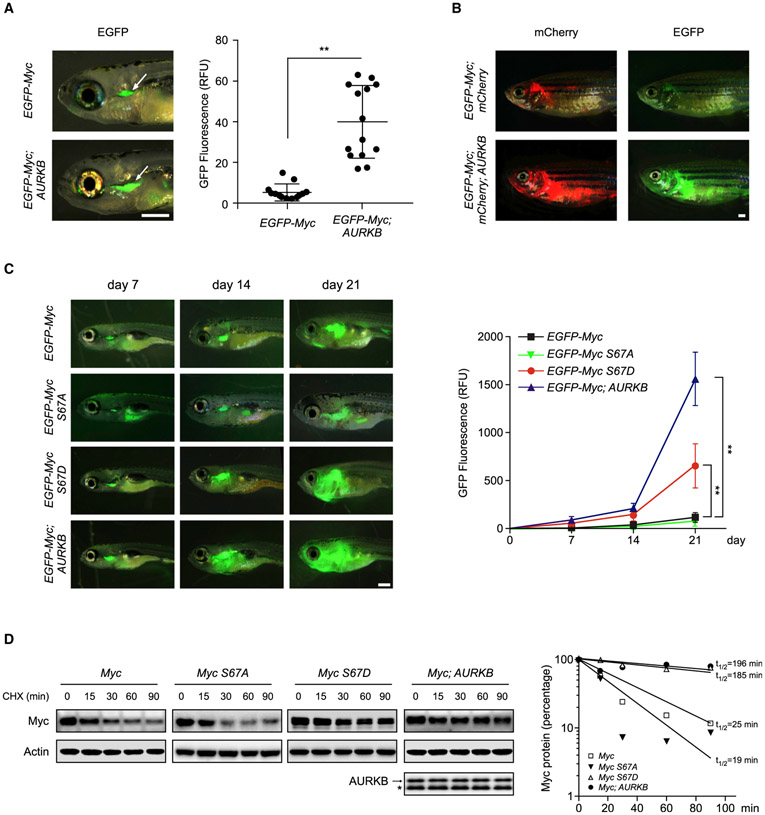
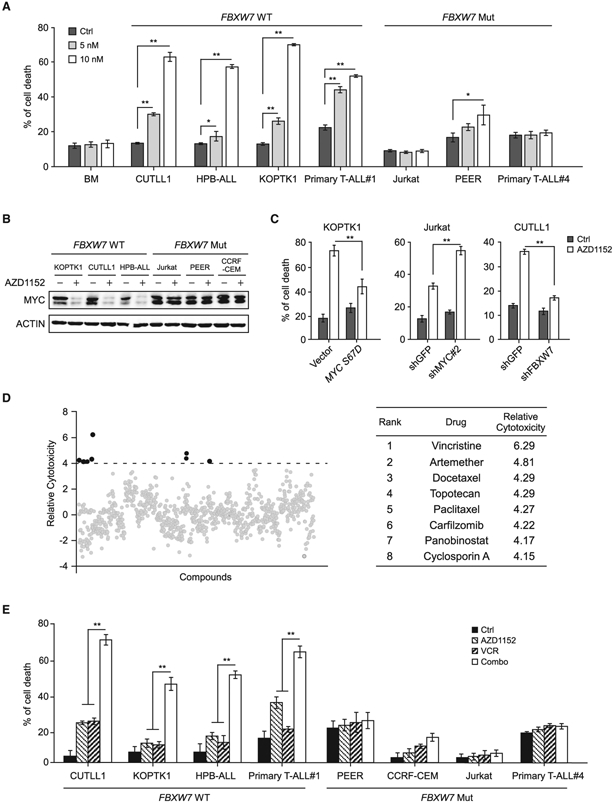
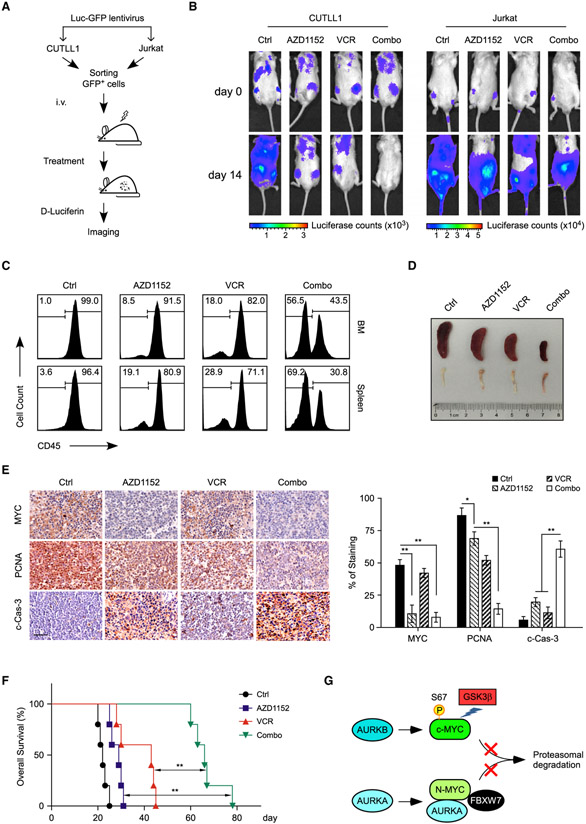
Deregulation of MYC plays an essential role in T cell acute lymphoblastic leukemia (T-ALL), yet the mechanisms underlying its deregulation remain elusive. Herein, we identify a molecular mechanism responsible for reciprocal activation between Aurora B kinase (AURKB) and MYC. AURKB directly phosphorylates MYC at serine 67, counteracting GSK3β-directed threonine 58 phosphorylation and subsequent FBXW7-mediated proteasomal degradation. Stabilized MYC, in concert with T cell acute lymphoblastic leukemia 1 (TAL1), directly activates AURKB transcription, constituting a positive feedforward loop that reinforces MYC-regulated oncogenic programs. Therefore, inhibitors of AURKB induce prominent MYC degradation concomitant with robust leukemia cell death. These findings reveal an AURKB-MYC regulatory circuit that underlies T cell leukemogenesis, and provide a rationale for therapeutic targeting of oncogenic MYC via AURKB inhibition.
Keywords: Aurora B kinase; FBXW7; MYC; T-ALL; patient-derived xenograft; phosphorylation; protein stability; zebrafish T-ALL model.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures

Similar articles
-
AURKB/CDC37 complex promotes clear cell renal cell carcinoma progression via phosphorylating MYC and constituting an AURKB/E2F1-positive feedforward loop.Cell Death Dis. 2024 Jun 18;15(6):427. doi: 10.1038/s41419-024-06827-y. Cell Death Dis. 2024. PMID: 38890303 Free PMC article.
-
Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival.Mol Cell. 2016 Nov 3;64(3):493-506. doi: 10.1016/j.molcel.2016.09.016. Epub 2016 Oct 20. Mol Cell. 2016. PMID: 27773673
-
Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state.Blood. 2010 Sep 2;116(9):1498-505. doi: 10.1182/blood-2009-11-251074. Epub 2010 Jun 2. Blood. 2010. PMID: 20519624 Free PMC article.
-
Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer.Molecules. 2021 Apr 1;26(7):1981. doi: 10.3390/molecules26071981. Molecules. 2021. PMID: 33915740 Free PMC article. Review.
-
Mitotic syndicates Aurora Kinase B (AURKB) and mitotic arrest deficient 2 like 2 (MAD2L2) in cohorts of DNA damage response (DDR) and tumorigenesis.Mutat Res Rev Mutat Res. 2021 Jan-Jun;787:108376. doi: 10.1016/j.mrrev.2021.108376. Epub 2021 Apr 24. Mutat Res Rev Mutat Res. 2021. PMID: 34083040 Review.
Cited by
-
A helicase-independent role of DHX15 promotes MYC stability and acute leukemia cell survival.iScience. 2023 Nov 23;27(1):108571. doi: 10.1016/j.isci.2023.108571. eCollection 2024 Jan 19. iScience. 2023. PMID: 38161423 Free PMC article.
-
HILPS, a long noncoding RNA essential for global oxygen sensing in humans.Sci Adv. 2023 Nov 24;9(47):eadi1867. doi: 10.1126/sciadv.adi1867. Epub 2023 Nov 22. Sci Adv. 2023. PMID: 37992175 Free PMC article.
-
MYC degradation via AURKB inhibition: a new brake in the path to T-ALL.Blood Sci. 2020 May 19;2(2):68-69. doi: 10.1097/BS9.0000000000000046. eCollection 2020 Apr. Blood Sci. 2020. PMID: 35402818 Free PMC article. No abstract available.
-
Aurora kinase B inhibition in small-cell lung cancer: BCL-2 as a potential therapeutic biomarker and combination target.Transl Lung Cancer Res. 2024 Mar 29;13(3):689-693. doi: 10.21037/tlcr-23-754. Epub 2024 Mar 15. Transl Lung Cancer Res. 2024. PMID: 38601438 Free PMC article. No abstract available.
-
Development of synthetic lethality in cancer: molecular and cellular classification.Signal Transduct Target Ther. 2020 Oct 19;5(1):241. doi: 10.1038/s41392-020-00358-6. Signal Transduct Target Ther. 2020. PMID: 33077733 Free PMC article. Review.
References
-
- Bachireddy P, Bendapudi PK, and Felsher DW (2005). Getting at MYC through RAS. Clin. Cancer Res 11, 4278–4281. - PubMed
-
- Bonnet M, Loosveld M, Montpellier B, Navarro J-M, Quilichini B, Picard, , Di Cristofaro J, Bagnis C, Fossat C, Hernandez L, et al. (2011). Posttranscriptional deregulation of MYC via PTEN constitutes a majoralternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood 117, 6650–6659. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
